Concepts
Lorikeet provides the user with a lot of different outputs for each genome and some of the information present in those outputs requires.
File types
FASTA
A FASTA file is a text based file used to represent either genomic nucleotide sequences or amino acids. It consists of
of headers (lines starting with >) and blocks of sequences immediately following the headers. Fasta files are the format
used for the input reference/MAGs that Lorikeet uses. The extension for such files is usually .fasta, .fa, or .fna.
For more info refer to the wikipedia article
FASTQ
FASTQ or the file format used to store data resulting from sequencing. The sequences present in FASTQ files represent short
genomic sequences of DNA. FASTQ files are used to build assemblies, MAG binnings, genomic coverage etc. You can provide
both paired end and unpared reads to Lorikeet, as well as short and long reads from a variety of different sequencing platforms.
The file extension for FASTQ files is generally .fastq, but often they have been compressed so the extension ends in .gz.
Compressed FASTQ files are accepted as input to Lorikeet so you do not have to uncompress them.
For more info refer to the wikipedia article
BAM/SAM
BAM and SAM (Sequence Alignment/Map) format files are the standard format for indicating the alignment start, end, and quality
of FASTQ files to FASTA files. BAM files are the binary format of SAM files, as such can not be read by conventional means.
When performing read mapping the output from the alignment tool will most likely be in SAM/ BAM files. Lorikeet produces
BAM files when supplied raw reads which can be stored using the --bam-file-cache-directory argument.
For more info refer to the SAM specification
BCF/VCF
The Variant Call Format (VCF) is a text file format used to store information about variation events found in a reference genome. The file consists of a series of information tags which specify the information that they hold, and then a series of lines representing the found variants. The variants will always contain the chromosome/contig they occur on, the position on that contig, the reference allele, and then any alternative alleles. The per sample depth of each allele is reported as individual grouped columns found after the INFO tags of each line. The way the information is stored can be rather confusing at first but there are a variety of python and R libraries which allow for easy parsing of VCF files like scikit-allel
Much like BAM and SAM files, VCF files also have a binary format called the BCF file format. BCF files allow for compressed
storage of bulky VCF files, and are easier for programs to deal with. To access the information stored within BCF files users
can make use of bcftools. For example, if a user wished to convert a BCF file generated by Lorikeet into a VCF file all they
would need to do is use the following command:
bcftools convert -O v -o output.vcf input.bcf
replace output.vcf with the desired output file name and input.bcf with the name of the BCF file you wish to convert.
For more info on BCF/VCF formats refer to the VCF specification and
for more info on bcftools refer to the BCFtools documentation
DOT
The graph description language DOT represents a series of nodes and edges for a given graph. Lorikeet produces a number of
these files during the genotype algorithm. The DOT files represent the links found between each variant group and the strength of
connection between them. They can be visualized using GraphViz
which has both an online and command line version.
Other
Lorikeet also produces a series of other file formats which you should be generally familiar with like txt and tsv files.
Among these however are the ANI files (consensus_ani.tsv, population_ani.tsv, and subpopulation_ani.tsv) which
pairwise matrices comparing the various ANI values between samples (non-diagonal cells) and the ANI values of a sample compared
to the reference (diagonal cells).
ANI
The average nucleotide identity (ANI) is a similarity index between a given pair of genomes that can be applicable to prokaryotic organisms independently of their G+C content. There is some debate about what ANI value should represent the cutoff for two genomes to represent the same species but values usually sit at either > 95% (or 0.95) or > 97% ANI (or 0.97). The ANI values typically produced by Lorikeet are typically much closer to 100% (or 1.0) than what is conventionally seen as we are measuring diversity at a much finer scale. Lorikeet reports ANI values between 0 and 1.0, which can be easily changed into a percentage if the user wishes to do so.
Consensus ANI
Consensus ANI, conANI, measures changes in the consensus allele seen between samples. The consensus allele being the allele
with the highest read depth in a given sample. The consensus allele is typically seen to represent the dominant allele within
community. Using only consensus ANI ignores any other diversity that may be present with in the community.
Population ANI
Population ANI, popANI, measures changes in the shared allelic composition of two communities. The population ANI between two
communities deviates away from 1.0 if those communities share no common allele at a given position.
Subpopulation ANI
Subpopulation ANI, subpopANI, also measures changes in the shared allelic composition of two communities. However,
subpopANI deviates away from 1.0 if those two communities do not share all the exact same alleles at a given position.
As such, it is much more sensitive changes in positions where more than two alleles are present in the community.
As an example, please refer to the following table displaying when each ANI measurement would deviated away from 1.0:
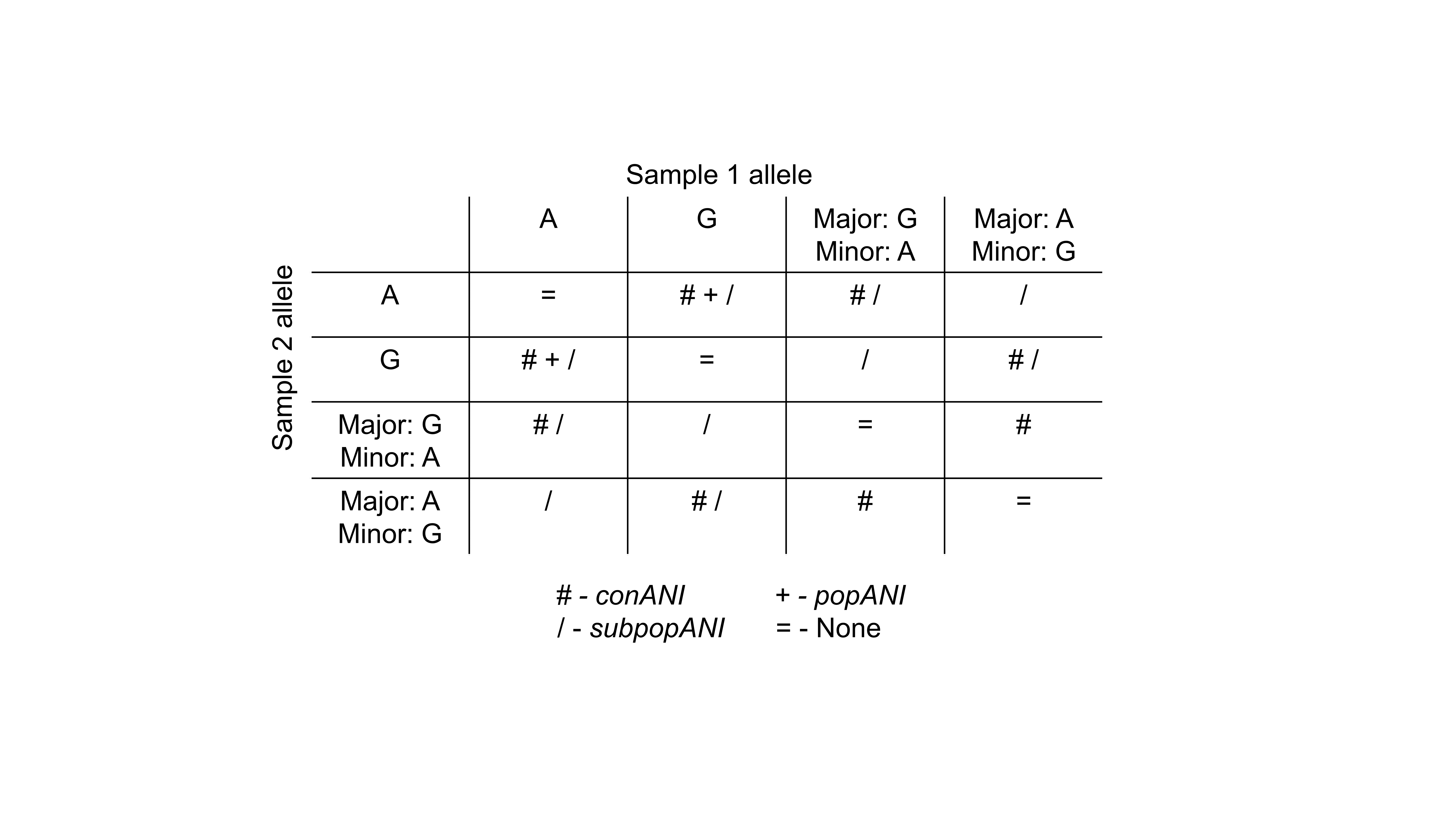
Strains
It has been said that "there is no universally accepted definition for the terms 'strain', 'variant', and 'isolate' in the virology community, and most virologists simply copy the usage of terms from others".1
For the purposes of Lorikeet, A strain is a genetic variant or subtype of a microorganism (e.g., a virus, bacterium or fungus). Within a microbial community, multiple strains of the same species can be present and can be measured by analyzing what variants are present in the community when compared to some reference genome. Ideally, this reference genome has been created from the microbial community being examined but this is not always possible.
Variant
Variants are the main tool with which we have to observe strains. They represent changes in our community compared to some reference genome. Variant can come in a variety of forms but the main three that Lorikeet identifies are:
SNPs
Single nucleotide polymorphisms (SNPs) represent single point changes against a reference. For example, the reference might contain an "A" at position 100 on contig 1, whilst a SNP at this position might suggest that a "G" is instead present here. SNPs are common and represent the vast majority of variants found within a community. If a SNP occurs within a coding region then it can be classified as either synonymous (No change in the encoded protein) or non-synonymous (changes the encoded protein).
INDELs
Insertions and Deletions (INDELs) represent a much more destructive form of variant. Insertions represent positions where bases have been "inserted" compared to the reference, while deletion represent "deleted" bases. INDELs can be small (only a couple of bases) or large (100s of bases), but even small INDELs can completely destroy a coding region.
MNVs
Multinucleotide variants (MNVs) sit somewhere between a SNP and an INDEL. They represent multiple nucleotide changes that are consistently seen with each other. MNVs can be a short chain of SNPs, or SNPs and INDELs.
Variant Group
A variant group is a set of variants that appear to cluster together across samples. This suggests that these variants are typically seen together in the same organism. A variant group does not usually represent a strain, as strains are built from multiple variant groups.
Powered by Doctave